深圳理工大学(筹)计算机科学与控制工程学院李金艳研究团队在国际知名学术期刊Nature Communications上发表了题为“Optimizing differential expression analysis for proteomics data via high-performing rules and ensemble inference”的研究文章。该研究为蛋白质组学数据的精准分析提供了新的视角和方法,对生物标记物和药物靶点的发现具有重要意义。
蛋白质组学数据的差异表达分析是生物医学领域中的关键技术,它能够帮助科学家准确检测特定表型的蛋白质。然而,由于分析流程中存在多种数据处理和分析工具的选择,确定最优的分析流程一直是一个挑战。为了解决这一问题,李金艳教授领导的研究团队对34,576种不同的差异分析流程进行了超大规模的性能评估。
通过频繁模式挖掘新技术,研究团队发现了一些能够提高差异表达分析性能的规则,并观察到这些规则具有跨平台的保守属性。此外,团队还构建了分类模型,证明了最优分析流程的可预测性。在此基础上,研究团队设计了一种集成推断方法,整合多个高性能差异分析流程的结果,以扩大差异蛋白质组的覆盖范围并消除分析流程结果间的不一致性。
集成推断方法不仅显著提高了差异表达蛋白鉴定的准确性,还促进了不同蛋白质量化方法之间的信息有效整合。例如,pAUC分数和G-均值分别获得了高达4.61%和11.14%的提高。研究团队还发现,使用不同蛋白质量化技术的分析流程之间具有更好的性能互补性,为设计更优异的差异表达分析方法提供了指导。
为了便于蛋白组学数据分析领域的研究者使用,研究团队构建了网络服务器OpDEA(http://www.ai4pro.tech:3838/)以及离线工具软件包,以协助研究者进行差异分析流程的选择和差异表达分析。
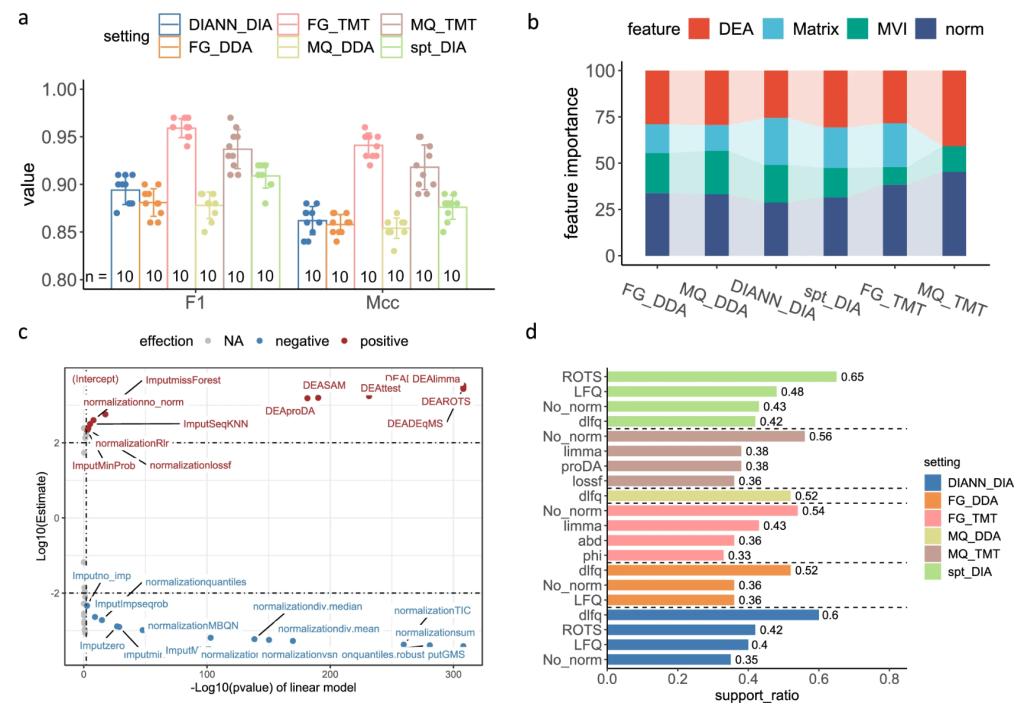
机器学习及数据挖掘技术分析差异表达分析流程的性能可预测性及保守性
阅读原文



